Cystisk fibrose er en arvelig sykdom som påvirker lungene og andre organer. I dag finnes ingen kur, men behandlingen har gjort stadige framskritt. Derfor kan personer med cystisk fibrose i dag leve betydelig lenger enn for noen år siden, med bedre livskvalitet.6
Cystisk fibrose er en arvelig sykdom som du arver fra dine foreldre. Globalt er det rundt 70 000–100 000 personer som har cystisk fibrose, og det oppstår rundt 1000 nye tilfeller hvert år.1,2 Cystisk fibrose er mest utbredt blant kaukasiere, og oppstår ved hver 2500 - 3000 levende fødsler. Cystisk fibrose oppstår sjeldnere hos andre etniske grupper.3
Personer med cystisk fibrose har et defekt protein som kontrollerer bevegelsen av salt og vann inn og ut av cellene.3 Dette defekte proteinet skyldes mutasjon eller unormalitet i et gen – cystic fibrose transmembran konduktans regulator-genet (CFTR).1,3
Cystisk fibrose påvirker cellene som produserer slim, svette og fordøyelsessafter. Mest påvirker cystisk fibrose lungene og fordøyelsessystemet, men det påvirker også bihuler, bukspyttkjertelen og forplantningsorganene (Figur 1).1 Under normale omstendigheter lager og utskiller disse organene væsker som er tyntflytende. Disse væskene er kjent som sekresjoner, og de fungerer som smøremidler. Hos individer med cystisk fibrose, blir disse sekresjonene tykke og seigere.3 På grunn av dette blokkerer de rør, kanaler og passasjer, spesielt i lungene og bukspyttkjertelen.4
Figur 1: Kroppsdeler som påvirkes av cystisk fibrose.4
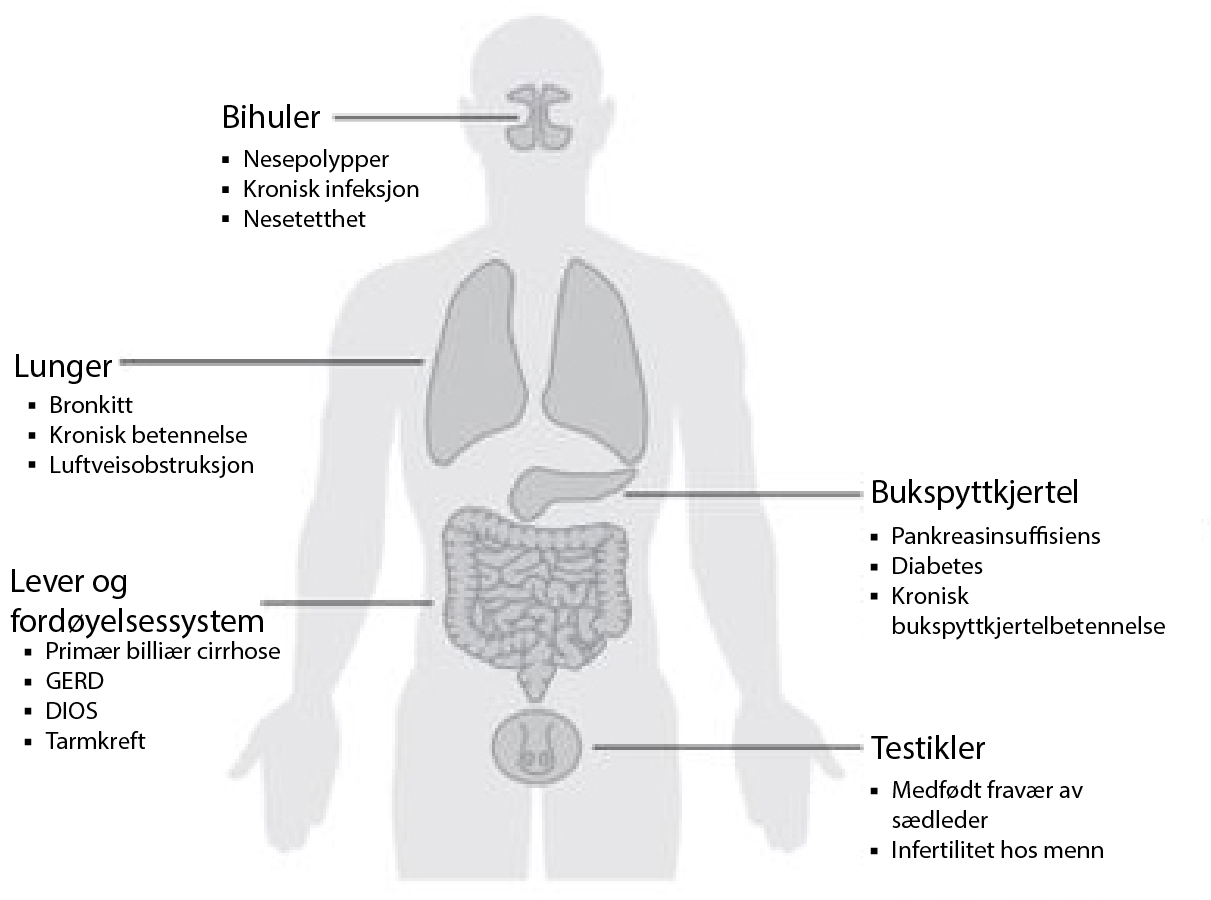
DIOS: Distalt intestinalt obstruksjonssyndrom (betyr blokkering av den distale delen av tynntarmen); GERD: Gastroøsofageal refluks (en fordøyelsessykdom som oppstår når sure fordøyelsessafter, eller mat og væske, kommer opp fra magen og inn i øsofagus og fører til halsbrann); GI: Gastrointestinal. Bronkietasi: En tilstand der lungenes luftveier blir skadet, noe som gjør det vanskelig å bli kvitt slim. Nesepolypper: En smertefri og godartet vekst av nesens innside eller bihulene. Medfødt fravær av vas deferens: Fravær av vas deferens fra fødselen av. Vas deferens er en del av det mannlige forplantningssystemet. Den leder sperm fra testiklene til penisen.
Hvem får cystisk fibrose?
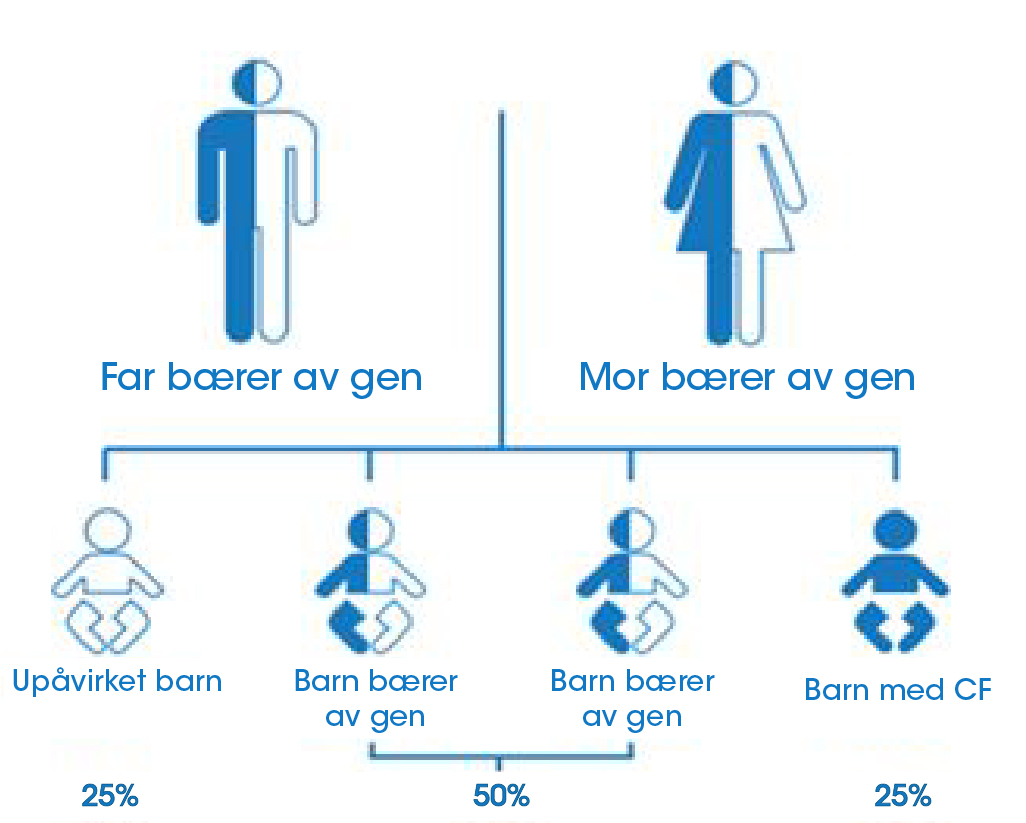
Cystisk fibrose er en genetisk sykdom. Den oppstår når et barn får to defekte gener, en fra hver forelder (figur 2). Når to personer som er bærere av et defekt gen får et barn sammen, er det:8
- 25% sjanse for at barnet vil bli født med cystisk fibrose8
- 50% sjanse for at barnet vil bli bærer av det defekte genet som er ansvarlig for cystisk fibrose4
- 25% sjanse for at barnet verken vil være bærer av eller bli født med cystisk fibrose8
Hvis cystisk fibrose er bekreftet, må også andre familiemedlemmer, inkludert søsken, undersøkes for sykdommen. Videre anbefales det at voksne slektninger også undersøkes, for å avdekke om de er bærere5
Figur 2: Hvem får cystisk fibrose?6
Tegn og symptomer på cystic fibrosis varierer avhengig av sykdommens alvorlighetsgrad.3 Personer med cystisk fibrose har salt svette.6 Andre vanlige tegn og symptomer på cystisk fibrose er at åndedrettssystemet og fordøyelsessystemet er påvirket.6 Voksne med cystisk fibrose har uvanlige symptomer. Dette kan være tilbakevendende episoder med bukspyttkjertelbetennelse (pankreatitt), tilbakevendende lungebetennelse og sterilitet.3
- Åndedrettstegn- og symptomer: Vedvarende hoste med spytt, hvesende pust, åndenød, gjentatte infeksjoner i lungene og betente neseganger6
- Fordøyelsetegn- og symptomer: Illeluktende og fettrik avføring; lite vektøkning og vekst, tarmblokkering, spesielt hos nyfødte (meconium ileus); og alvorlig forstoppelse5
Steatore og lite vektøkning skyldes eksokrin pankreasinsuffisiens (EPI). Cystisk fibrose er en av hovedårsakene til EPI; rundt 80–90% av personer med cystic fibrose utvikler EPI. De fleste pasienter med cystisk fibrose blir diagnostisert med EPI i løpet av sitt første leveår. Eksokrin pankreasinsuffisiens anses som en hovedårsak til dårlig opptak og feilernæring hos pasienter med cystisk fibrose. Det er derfor vanlig å få tilskudd av enzymer Pankreatin, for å bedre ernæringen.7
Cystisk fibrose er en sykdom som forkorter levetiden. Med dagens fremskritt innen diagnose og behandling, har overlevelsesraten blitt bedre. Personer med cystisk fibrose kan nå leve i over 40 år.3,6 Dette er en fordobling i forhold til leveralder for 40 år siden.3
Selv om pasienter med cystisk fibrose trenger daglig omsorg og noen ganger har vanskeligheter med å leve et normalt liv, så kan de vanligvis gå på skole og jobb. De har også ofte bedre livskvalitet sammenlignet med personer med cystisk fibrose i tidligere tiår.6
-
Referanser
- Schneider-Futschik EK. Beyond cystic fibrosis transmembrane conductance regulator therapy: A perspective on gene therapy and small molecule treatment for cystic fibrosis. Gene Therapy. 2019;26:354–362.
- Rafeeq MM, Murad HAS. Cystic fibrosis: Current therapeutic targets and future approaches. J Transl Med.2017;15:84–92.
- Karen Helene Ørstavik, Jaran Apold, Genterapi ved monogent arvelige sykdommer, Tidsskr Nor Lægeforen , 121: 349-50, Utgave 30 januar, 2001
- Rey MM, Bonk MP, Hadjiliadis D. Cystic fibrosis: Emerging understanding and therapies. Annu Rev Med. 2019;70:197–210.
- Davies JC, Alton EWFW, Bush A. Cystic fibrosis. BMJ. 2007;335:1255–1259. Kashirskaya NY, Kapranov NI, Sander-Struckmeier S, et al. Safety and efficacy of Creon® Micro in children with exocrine pancreatic insufficiency due to cystic fibrosis. J Cyst Fibros. 2015;14:275–281.
- Pediatriveiledere fra Norsk barnelegeforening.: Generell veileder, øvre og nedre luftveier, Cystisk fibrose 7,14: Revidert 2018
- Kashirskaya NY, Kapranov NI, Sander-Struckmeier S, et al. Safety and efficacy of Creon® Micro in children with exocrine pancreatic insufficiency due to cystic fibrosis. J Cyst Fibros. 2015;14:275–281.
- Oslo Universitetssykehus: Cystisk fibrose - årsaker og arvegang, Publisert 21.10.2016

Hva er eksokrin pankreasinsuffisiens?
Langvarige diaréer, vekttap og generelle fordøyelsesproblemer kan skyldes mangel på fordøyelsesenzymer. Dette kalles eksokrin pankreasinsuffisiens (EPI).

Behandling av EPI
Å gjenopprette normal fordøyelse er målet med behandlingen av EPI. Denne behandlingen består ofte av livsstil og kostendringer samt å erstatte manglende enzymer fra bukspyttkjertelen for å bedre fordøyelsen av maten.