Cystisk fibrose forårsakes på grunn av et defekt gen. Man kan ikke få CF på samme måte som en forkjølelse eller hoste, og det kan heller ikke utvikle seg som kreft. CF er en genetisk sykdom – hvis du ikke er født med cystisk fibrose, så vil du aldri få det, men du kan være en bærer av genet som gir sykdommen.1
Cystisk fibrose er en genetisk eller arvelig sykdom som påvirker både kroppens celler, vev og kjertler som lager slim og svette. Cystisk fibrose påvirker forskjellige kroppsdeler og personer med denne tilstanden kan oppleve en rekke symptomer avhengig av hvilket organ som er påvirket. Behandlingen av cystisk fibrose har som mål å forbedre symptomer og forhindre andre komplikasjoner.1
Hva forårsaker cystisk fibrose
Cystisk fibrose forårsakes av feil eller mutasjoner i et gen som heter cystisk fibrose transmembran konduktans regulator-gen (CFTT). Dette CFTR-genet er ansvarlig for å lage CRFT-proteinet – dette proteinet er tilstede i svettekjertler, samt i alle organer som lager slim, som lunger, lever, bukspyttkjertel og tarmene. Under normale forhold er disse slimsekresjonene tynne og fungerer som smøremidler.1
Hva gjør CFTR-proteinet ?
CFTR-proteinet kontrollerer bevegelsen av salt fra innsiden til utsiden av cellen. Hos personer med cystisk fibrose fører det defekte genet til at proteinet ikke fungerer som det skal. Dette forstyrrer igjen bevegelsen av salt og vann. På grunn av denne forstyrrelsen i transport av salt og vann, er svetten som kommer ut av svettekjertlene saltere enn hos personer uten cystisk fibrose. I tillegg er det mindre vann i slimet, noe som gjør det tykt og seigere. Det tykke slimet skaper blokkeringer i lungene og fordøyelsessystemet.1 Det er rundt 2000 mutasjoner i CFTR genet. Alvorlighetsgraden av tilstanden varierer avhengig av typen genmutasjoner.3
Det er rundt 2000 mutasjoner i CFTR genet. Alvorlighetsgraden av tilstanden varierer avhengig av typen genmutasjoner.3,4
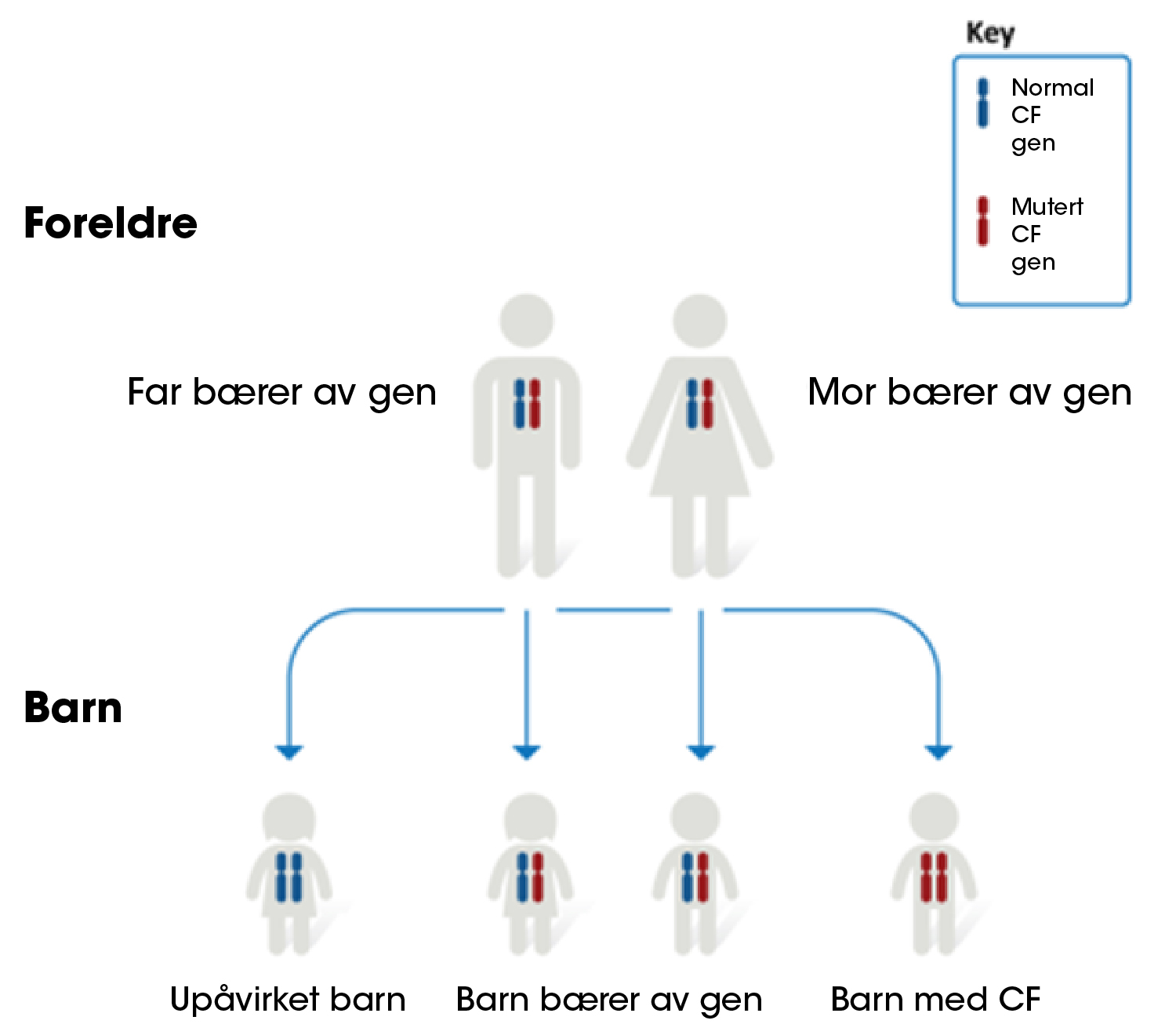
Cystisk fibrose er en genetisk sykdom, den oppstår når et barn får to defekte gener, en fra hver foreldre (figur 1). Når to personer som er bærere av en genfeil som kan forårsake cystisk fibrose får et barn sammen, er det:2,4
- 25% sjanse for at barnet vil bli født med cystisk fibrose2
- 50% sjanse for at barnet vil bli bærer av genfeilen som kan forårsake cystisk fibrose. Barn som arver bare en kopi, vil ikke utvikle cystisk fibrose. De vil imidlertid være bærere, og kan muligens videreføre genet til sine egne barn.2
- 25% sjanse for at barnet verken vil være bærer av eller bli født med cystisk fibrose2
Figur 1: Hvem får cystisk fibrose?1
Personer med familiehistorikk med cystisk fibrose eller personer som tilhører en spesifikk etnisitet kan ha en økt risiko for cystisk fibrose.1
- Familiehistorikk: Cystisk fibrose er en arvelig sykdom, den ligger i familien.1
- Etnisitet: Cystisk fibrose kan påvirke personer fra alle raser, men er vanligst blant personer fra Nord-Euorpa og mindre vanlig blant personer fra Latin-Amerika og Afro-Amerikanere.1 CF er relativt uvanlig blant amerikanere med asiatisk opphav.
Gentesting kan utføres for å se etter bærere, og for å screene pårørende til personer som har cystisk fibrose.1
Undersøkelse av bærere for å oppdage CFTR-genmutasjoner
- Gentesting kan fortelle om en person er bærer av en mutasjon av CFTR-genet. Dette kalles bærertesting. Standardtesten ser etter de 23 vanligste gendefektene som forårsaker genmutasjoner.1
- Søsken til en person som er diagnostisert med cystisk fibrose bør sjekke seg for cystisk fibrose, uansett om vedkommende har symptomer eller ikke. Andre slektninger som søskenbarn og halvsøsken, bør testes hvis de har symptomer.1
Nyfødt undersøkelse
- Det er svært viktig å diagnostisere cystisk fibrose så tidlig som mulig.4
- Tidlig diagnostisering bidrar til å forhindre komplikasjoner og forbedrer barnets vekst og næringsstatus.4
- Undersøkelse for cystisk fibrose hos nyfødte utføres i løpet av en babys første to til tre levedager. Hvis testresultatene skulle vise seg å være positive, må diagnosen bekreftes med videre testing.4
-
Referanser
- Pediatriveiledere fra Norsk barnelegeforening.: Generell veileder, øvre og nedre luftveier, 7.14 Cystisk fibrose : Sist oppdatert: 28.09.2023.
- Oslo Universitetssykehus: Cystisk fibrose - årsaker og arvegang, Sist oppdatert: 13.07.2023.
- Schneider-Futschik EK. Beyond cystic fibrosis transmembrane conductance regulator therapy: A perspective on gene therapy and small molecule treatment for cystic fibrosis. Gene Therapy. 2019;26:354–362.
- Davies JC, Alton EWFW, Bush A. Cystic fibrosis. BMJ. 2007;335:1255–1259.

Årsaker til EPI
Ekoskrin pankreasinsuffisiens (EPI) kan også oppstå som en følge av andre sykdommer i bukspyttkjertelen som kronisk pankreatitt, diabetes, eller etter kirurgi i magen eller bukspyttkjertelen.

Behandling av EPI
Å gjenopprette normal fordøyelse er målet med behandlingen av EPI. Denne behandlingen består ofte av livsstil og kostendringer samt å erstatte manglende enzymer fra bukspyttkjertelen for å bedre fordøyelsen av maten.